

Síndrome de Vogt-Koyanagi-Harada
Paciente mujer hispanoamericana de 14 años que acude al servicio de urgencias por la persistencia de fiebre y aumento de malestar general, con síndrome febril de predominio vespertino de hasta 39,9ºC, de aproximadamente 15 días de evolución.
Dres. García Giralda M (ORL), Alfaro Juárez A (Oftalmología). Hospital Comarcal de Baza (Granada)
Publicación 01-08-2017
El síndrome de Vogt-Koyanagi-Harada es una enfermedad sistémica autoinmune, caracterizada por la aparición de uveítis, hipoacusia, alopecia, encefalitis y vitíligo. La reacción inflamatoria provocada por la alteración del sistema inmunitario afecta a diversos órganos ricos en pigmentos (úvea, retina y algunos pares craneales causantes de la pérdida de audición).
El primero en describirlo fue un médico árabe en torno al año 940 a. C. En 1906, Vogt, y en 1929, Koyanagi, describieron diversas modalidades del síndrome en pacientes que presentaban iridociclitis, meningoencefalitis asociada a vitíligo, poliosis (pestañas blancas), alopecia e hipoacusia. Por la misma época (1926) Harada describe el cuadro de un paciente con uveítis bilateral asociada a encefalitis, adoptándose finalmente en 1951 la nomenclatura actual. La enfermedad suele comenzar con un episodio febril seguido de uveítis bilateral asociada a coroiditis y neuritis óptica. Puede acompañarse de hipoacusia neurosensorial y tinnitus, vitíligo, poliosis y alopecia, y en algunos casos incluso meningitis o encefalitis. Afecta más frecuentemente a personas de 20-50 años, de raza asiática e hispanoamericana y a mujeres el doble que a hombres.
Descripción del caso clínico
Paciente mujer hispanoamericana de 14 años que acude al servicio de urgencias por la persistencia de fiebre y aumento de malestar general, con síndrome febril de predominio vespertino de hasta 39,9ºC, de aproximadamente 15 días de evolución. Asocia desde el inicio del cuadro, dolor abdominal epigástrico, por lo que en visitas anteriores al servicio de urgencias se le ha solicitado ecografía abdominal y se han sacado serologías de virus, siendo todas ellas negativas. En las analíticas evolutivas llama la atención anemia normocítica normocrómica, linfopenia, así como plaquetopenia, alteraciones en coagulación y aumento progresivo de la PCR, por lo que se ingresa en el servicio de medicina Interna ante la sospecha de síndrome linfoproliferativo.
- Serología sistémicas/AI/otras: ANA, pANCA, cANCA, FR y CCP: negativos.ECA: normal (33.4 U/L; VR 20-70).
- Anticoagulante Lúpico y Ac antifosfolípido: negativos.Transglutaminasa negativo. Inmunoglobulinas normales.
- Cadenas ligeras normales. HLA-DRB1, HLA-DRB4, HLA-B*27: negativo, HLA-B*51: negativo.
- Serología infecciosas: VHB, VHC, VIH, CMV, VEB, VVZ, V sarampión, virus de la parotiditis, parvovirus virus toscana: negativos. Borrelia burgdorferi, Brucella, Legionella pneumophila, Mycoplasma pneumoniae, Coxiella burnetii, Chlamydophila pneumoniae: negativos. Bartonella henselae: negativa.Trypanosoma cruzi: negativo. RPR: negativo.
- Hemocultivos: negativos.
- Cultivo esputo: BK negativo. Cultivo negativo. Virus de la influenza A y B negativo, Virus respiratorio sincitial: negativo.
- Urocultivo: negativo.
- Mantoux: negativo.
- Antigenuria Legionella, Neumococo y Lehismania negativos.
- Parásitos en heces: negativo. Estudio de LCR.
- Microbiología: cultivo bacterias negativo. VHS, VVZ, Toscana, enterovirus, parotiditis y parechovirus: negativos.
- Anatomía patológica: extendidos hipercelulares constituidos por leucocitos polimorfos fundamentalmente neutrófilos con presencia de linfomonocitos, sugerente de proceso infeccioso agudo.
- Rx de tórax: infiltrado intersticial bilateral, micronodular (miliar) de confluencia mayor en lóbulo derecho.
- Ecografía de abdomen normal.
- Ecocardiografía tt: derrame pericárdico posterior y sobre VD de grado ligero sin compromiso hemodinámico. Derrame pleural ligero. No evidencia de CIA ni CIV.
- TC con contraste I.V. de abdomen y de tórax: adenopatías axilares bilaterales y mediastínicas de 12 mm. No lesión pleuroparenquimatosa, excepto bronquiectasias bilaterales. Aumento de tejido linfoide a nivel de aa. pulmonar derecha. Hepatomegalia, por aumento de LHI.
- RM cráneo: normal.
- Fibrobroncoscopia, (al mes de ingreso) bajo anestesia general por dificultades en el control y tolerancia de pruebas diagnósticas.
- Tomografía por Emisión de Positrones (PET), mediante rastreo tomográfico desde cráneo hasta región inguinal: normal.
Evolución y curso clínico
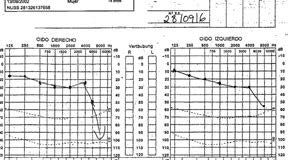
Evolutivamente desarrolla síndrome uveomeníngeo; meningitis linfocitaria con uveítis bilateral anterior granulomatosa y sordera neurosensorial bilateral con pérdida del 50% de la agudeza auditiva (figura 1).
Ante la sospecha de enfermedad autoinmune, Vogt-Koyanagi-Harada (VKH) tipo II, por los problemas de visión y audición (no tiene vitíligo pero sí una pestaña blanca, poliosis) se decide tratamiento con corticoides, mejorando la clínica rápidamente en una semana, la audiometría casi se normaliza (figura 2) y mejora muy ostensiblemente del síndrome uveomeníngeo, dándole el alta.
El tratamiento se realiza con: prednisona 40 mg, vía oral al día, durante una semana; para a continuación reducir la dosis 5 mg cada semana (4 semanas), luego cada cuatro semanas y, finalmente, seguir con 2,5 mg de mantenimiento, 6-12 meses hasta revisión. Tratamiento ocular: dexametasona colirio 1mg/ ml; 1 gota cada 4 horas durante 7 días, después 1 gota cada 6 horas hasta revisión por oftalmología. Además, para la anemia, hierro (II) sulfato 325 mg, durante un mes. Revisión en 3 meses.
Discusión
La enfermedad de VKH consiste típicamente en 4 fases. La primera fase o prodrómica se caracteriza por manifestaciones neurológicas y auditivas; una segunda fase uveítica aguda con coroiditis difusa, desprendimientos exudativos de retina y papilitis; la tercera fase, crónica, caracterizada por despigmentación de diferentes estructuras tanto oculares como tegumentarias; y una cuarta fase, crónica recurrente.
Los hallazgos del examen clínico dependerán del estadio clínico de la enfermedad. Se clasifica, de acuerdo con el compromiso orgánico, en:
- Tipo I o probable: afectación ocular sin compromiso neurológico o dérmico.
- Tipo II o incompleto: hallazgos oculares y al menos una manifestación neurológica o dérmica.
- Tipo III o completo: signos oculares con dos o más manifestaciones neurológicas o dérmicas.
No existen tests específicos para confirmar el diagnóstico, siendo este fundamentalmente clínico. Para ello se utilizan los criterios establecidos por la Sociedad Americana de Uveítis.
El diagnóstico se realiza mediante el examen ocular completo. Se indica angiografía, pruebas de laboratorio (hemograma, química sanguínea y estudios inmunológicos) y estudios de imagen y del líquido cefalorraquídeo de ser posible.
Los pacientes con síndrome de VKH completo deben presentar todos los criterios clínicos establecidos por la Sociedad Americana de Uveítis en 1978 y de la revisión realizada en 1999 por el Comité Internacional de VKH.
Los del tipo incompleto deben cumplir los primeros tres criterios además de presentar alteraciones neurológicas, auditivas o cutáneas y los del tipo probable sólo presentan alteraciones oculares.
Es importante el diagnóstico diferencial con otras enfermedades que presentan desprendimiento de retina exudativo bilateral como el síndrome de infusión uveal, tumores (linfoma primario de células B), causas inflamatorias como escleritis posterior y vasculitis e infeccionas como sífilis y toxoplasmosis y con la sarcoidosis.
Aunque no existe un tratamiento específico para la enfermedad de VKH, los síntomas, generalmente, pueden ser controlados por los corticosteroides y agentes citotóxicos.
El pronóstico visual de los pacientes es, generalmente, bueno si el diagnóstico es temprano y se prescribe un tratamiento adecuado de forma agresiva y mantenida en el tiempo. El síndrome VKH, a pesar de ser una enfermedad poco frecuente, presenta complicaciones importantes como la pérdida de la visión en pacientes jóvenes, por lo que su diagnóstico temprano se vuelve esencial para un mejor pronóstico.

Archivo: PDF Tamaño: 0.73
Bibliografia
- Zúñiga José H, Rodas Óscar M, Morales Ismael G. Síndrome de Vogt Koyanagi Harada, a propósito de un caso. ImedPub Journals.Archivos de medicina.ISSN 1698-9465, 2016, vol.12 no 1:1.
- Gil Hernández MA, Abreu Reyes P, Hernández Brito A, Castellano Solanes J, Herrera Pinero R. Severe evolution of VogtKoyanagi-Harada disease. Archivos de la Sociedad Canaria de Oftalmología. 1986-1998.
- Matsuda H, Sugiura S. Ultraestructural changes of the melanocyte in VKH sindrome and sympathetic ophtalmia. Jpn J Ophthalmol 1971; 15: 69-80.
- Norose K, Yano A, Wang X, et al. Dominance of activated T cells and interleukin in aqueous humor in VKH syndrome. Invest Ophthalmol Vis Sci 1994; 35: 33-39.
- Otani S, Sakurai T, Yamamoto K, Fujita T, Matsuzaki Y, Goto Y. Frequent immune response to a melanocyte specific protein KUMEL-1 in patients with Vogt-Koyanagi-Harada disease. Br JOphthalmol 2006; 90: 773-777.
- Duke Elder S, Perkins ES. Vogt-KoyanagiHarada syndrome. In Duke Elder (ed): Diseases of the uveal Tract. Mosby 1996; Vol 9: 373-383.
- Fishman R. Cerebrospinal fluid in diseases of the nervous system. Edit. Mills L.E. Ed. W.B. Saunders Co. Philadelphia. Pennsylvania 19106. Second Edition. pp.291, 1992.
- De-Domingo B, Blanco MJ, Rodríguez-Cid MJ, Piñeiro A, Mera P, Capeáns MC. Vogt-KoyanagiHarada Syndrome. Archivo de la Sociedad Española de Oftalmología 2008; 83: 385-390.
Si quieres participar enviando casos clínicos, imágenes clínicas comentadas o formación médica, puede enviárnolos a gaesmedica@cpp-proyectos.com.
PDF Tamaño: 205 kb