

Macroadenoma hipofisario productor de hormona del crecimiento
Mujer de 43 años de edad, con antecedentes de HTA, dislipemia, migrañas diagnosticadas hace 2-3 años. No alergias medicamentosas. Había consultado una semana antes por cefalea intensa y cervicalgia que relacionó con las migrañas que padece.
Dres. Arjona Montilla C, García Giralda M, Sánchez Rozas JA, Pérez Villoslada J. Hospital Comarcal de Baza (Granada).
Publicación 01-09-2017
Descripción del caso clínico
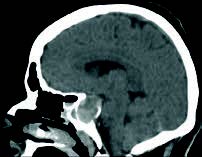
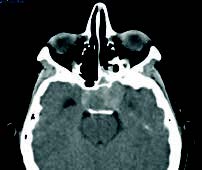
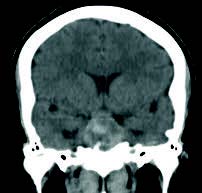
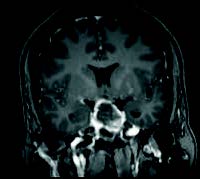
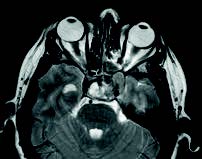
Mujer de 43 años de edad, con antecedentes de HTA. Dislipemia. Migrañas diagnosticadas hace 2-3 años. No alergias medicamentosas. Había consultado una semana antes por cefalea intensa y cervicalgia, que relacionó con las migrañas que habitualmente padece, consultó por visión borrosa, de predominio en ojo derecho. Fue diagnosticada de sinusitis y se inició tratamiento antibiótico. Desde entonces, refiere que no ve con el ojo derecho y la visión con el ojo izquierdo es borrosa. Acude al servicio de urgencias del hospital, donde, tras la realización de un TAC craneal, se aprecia tumoración de la silla turca que invade seno esfenoidal sugerente de macroadenoma hipofisario.
Es derivada al servicio de neurocirugía del hospital de referencia donde se practica RM craneal, que muestra: imágenes compatibles con voluminoso macroadenoma hipofisario, de 34 T x 34 AP x 25 CC mm, invasivo, principalmente, hacia celdillas etmoidales posteriores izquierdas y hacia el seno cavernoso izquierdo, donde rechaza hacia atrás y engloba circunferencialmente a la ACII. En el interior de la tumoración existen imágenes de alta susceptibilidad magnética que sugieren sangrado agudosubagudo. Ocupa la cisterna supraselar donde comprime considerablemente el quiasma y porción prequiasmática de nervios ópticos, sobre todo el derecho. Cutis verticis gyrata que permite considerar como primera posibilidad macroadenoma productor de GH (hormona de crecimiento).
El paciente es intervenido con carácter urgente por compromiso visual por el servicio de ORL vía endoscópica transesfenoidal. Anatomía patológica: neoplasia epitelial formada por células semejantes a las adeno-hipofisarias, con red vascular prominente que muestra amplias áreas de infarto. Con la tinción para reticulina se observa pérdida del patrón característico de la adenohipófisis normal. Las células neoplásicas son positivas para citoqueratina (CAM 5.2), con un patrón predominantemente en localización paranuclear, y para cromogranina. Además, se ha observado positividad inmunohistoquímica heterogénea y focal para GH. El índice proliferativo, determinado con la tinción para Ki67, aunque es difícil de determinar por la necrosis presente, es de un 1,58%, aproximadamente. Conclusión: adenoma productor de GH pobremente granulado con infarto masivo.
Discusión
Los endocrinólogos clínicos utilizan a menudo la clasificación funcional de los adenomas hipofisarios y definen estos tumores basados en su actividad hormonal en vivo. Un examen retrospectivo de la literatura del adenoma hipofisario indica que los prolactinomas son, fácilmente, la forma más común de adenoma hipofisario, según lo determinan los criterios inmunohistoquímicos. Le siguen en orden descendente, los tumores que secretan la hormona adrenocorticotrópica (ACTH), la hormona luteinizante (LH) y la hormona estimulante de la tiroides (TSH).
Los adenomas hipofisarios inactivos funcionales, sin embargo, comprenden aproximadamente de 30 a 35% de los tumores de hipófisis en la mayoría de las series y son el tipo más común de macroadenoma. En los productores de GH, la curación completa es posible solamente tras un tratamiento quirúrgico. La evaluación se realizará a los 3-6 meses: concentración de GH <46 pmol/l (<1 µg/l) en la prueba de tolerancia oral a la glucosa (los valores muestran una mínima concentración sérica de GH obtenida en cualquier punto de la prueba) y concentración de IGF-1 dentro del rango normal para el sexo y la edad.
Se considera enfermedad controlada (durante el tratamiento con análogo de somatostatina): si se mantiene la concentración sérica de GH <46 pmol/l (<1 µg/l) y disminuye la concentración de IGF-1 hasta el rango normal para el sexo y la edad.
Cirugía transesfenoidal es el método de elección (tras la preparación con un análogo de somatostatina de acción prolongada). Puede conseguir la curación completa.
El tratamiento farmacológico se realiza con análogos de somatostatina de acción prolongada:
Octreotide dosis inicial de 20 mg IM 1 × mes; si a los 3 meses la concentración de IGF-1 no vuelve al rango normal → aumentar la dosis hasta 30 mg IM 1 × mes, o lanreotida dosis de 60-120 mg VSc cada 4 semanas; si el tratamiento es eficaz, administrar una dosis de 120 mg VS cada 6 u 8 semanas.
- Antes de la resección del macroadenoma, especialmente en caso de infiltración de los tejidos adyacentes. En la mayoría de los casos se logra disminuir y en ~50% normalizar la concentración de GH, así como disminuir el tamaño del tumor y cambiar su consistencia, lo que facilita la resección completa.
- Tras la resección del macroadenoma, si la cirugía no ha sido eficaz.
- En pacientes que no han sido sometidos a cirugía, por contraindicaciones o falta de acuerdo con paciente.
En caso de ineficacia de los análogos de somatostatina → se debe añadir un fármaco dopaminérgico, antagonista de la hormona de crecimiento (pegvisomant), reoperar el tumor o, en última instancia, considerar la radioterapia.
La radioterapia estereotáxica o conformada puede servir de tratamiento complementario en caso de ineficacia del tratamiento quirúrgico y farmacológico.
En pacientes no tratados con acromegalia, la mortalidad debida a enfermedades del sistema cardiovascular, respiratorio y a enfermedades neoplásicas es 2-4 veces mayor que en la población general.

Archivo: PDF Tamaño: 0.78
Bibliografia
- Moreno B, Obiols G, Páramo C, Zugasti A. Guía clínica del manejo del prolactinoma y otros estados de hiperprolactinemia. Endocrinología y Nutrición 2005 (52): 9-18.
- Gillam MP, Molitch ME, Lombardi G, Colao A. Advances in the treatment of prolactinomas. Endocr Rev 2006; 27: 485-534.
Suplemento de Acromegalia. Endocrinología y Nutrición 2005; 52 (supl 3): 1-57.
Diagnosis and treatment of acromegaly and its complications: consensus guidelines. J Endocrinol Invest. 2005; 28 (supl 11): 43-7. - Lucas T, Catalá M. Guía clínica del diagnóstico y tratamiento de la acromegalia. Endocrinología y Nutrición 2005 (52): 9-18.
- Lucas T, Páramo C, Torres E, Catalá M, Gilsanz A, Moreno B, Obiols G, Picó A, Tortosa F, Varela C, Zugasti A, Villabona C. Guía clínica del diagnóstico y tratamiento del incidentaloma hipofisario. Endocrinología y Nutrición 2005; 52 (supl 3): 9-13.
- Gilsanz A, Moreno B, Obiols G, Zugasti A, Catalá M, Lucas T, Páramo C, Picó A, Torres E, Tortosa F, Varela C, Villabona C. Guía clínica del diagnóstico y tratamiento de los tumores hipofisarios no funcionantes y gonadotropinomas. Endocrinología y Nutrición 2005; 52 (supl3): 3-19.
Si quieres participar enviando casos clínicos, imágenes clínicas comentadas o formación médica, puede enviárnolos a gaesmedica@cpp-proyectos.com.
PDF Tamaño: 205 kb